Введение
Расстройства аутистического спектра (РАС) — это сохраняющиеся на протяжении всей жизни очень разнообразные нарушения психического развития, клиническими проявлениями которых являются нарушение социального взаимодействия и коммуникации, ограниченные интересы и стереотипное поведение [Diagnostic and statistical, 2013]. Распространенность РАС увеличивается; следовательно, необходимо определить ключевые механизмы его этиологии, которые помогут облегчить идентификацию прогностических и диагностических маркеров и помочь в разработке терапевтических методик. Существует множество этиопатологических механизмов, связанных с аутизмом; наиболее признанные из них включают системную иммунную активацию и эксайтотоксичность. Считается, что хроническое воспаление является отличительной особенностью многих нарушений психического развития [Mead, 2015; Pardo, 2005]; согласно исследованиям, у значительного количества пациентов с РАС наблюдается дисфункция иммунной системы [Edmiston, 2017; Mostafa, 2012]. Нейровоспаление было обнаружено в посмертных препаратах мозга молодых и пожилых людей с аутизмом [Gegelashvili, 2014; Pardo, 2005; Vargas, 2005].
В настоящий момент появляется все больше доказательств того, что генетические и средовые факторы риска при аутизме приводят к нарушению баланса между возбуждающей глутамат-опосредованной и ингибирующей ГАМК-опосредованной нейротрансмиссией; это может помочь определить цели терапии при данном расстройстве [Borisova, 2018; Nelson, 2015; Rubenstein, 2003; Sano, 2009].
Гипотеза, согласно которой ГАМК может быть выбрана в качестве мишени, подходящей для терапии аутизма, подтверждается достаточно высокой распространенностью эпилепсии у пациентов с РАС, а также частым присутствием эпилептиформной активности, наблюдаемой на электроэнцефалограммах (ЭЭГ) пациентов с РАС [Robertson , 2015].
Более того, после блокирования материнского окситоцина, играющего важную роль в раннем постнатальном переходе ГАМК-эргического сигнального пути от возбуждения к торможению, у мышей наблюдали появление электрофизиологических и поведенческих особенностей, сходных с проявлениями аутизма [Tyzio, 2016; Tyzio, 2014].
Эти данные позволяют считать модель дисбаланса возбуждения и торможения одной из ведущих гипотез, объясняющих этиологию аутизма. Следовательно, точная и интегрированная корректировка обмена веществ в тканях мозга посредством регуляции уровня глутаминовой кислоты и ГАМК может стать стратегией лечения аутизма.
У нас пока нет четкого механизма действий в отношении введения добавок ГАМК, и нам еще предстоит получить полную информацию о роли ГАМК в изменении поведения, а также о возможности ее прохождения гематоэнцефалического барьера (ГЭБ) у людей. Однако согласно данным многочисленных клинических исследований, ГАМК обладает терапевтическим эффектом для мозга [Boonstra, 2015; Fiorentino, 2016; Leonte, 2018; Reuter, 2017].
Связь метаболизма глутамата и ГАМК
Широко известно, что метаболизмы глутамата и ГАМК тесно связаны, поэтому изменение любого промежуточного метаболита может оказывать влияние на оба нейротрансмиттера. Глутаминаза — это фермент, катализирующий превращение глутамата в глутамин, при этом глутамин может либо храниться в астроцитах, либо преобразовываться в глутамат и в глутаматергических, и в ГАМК-эргических нейронах [Rowley, 2012]. Между синаптическими событиями нормальные уровни глутамата и ГАМК остаются низкими за счет постоянного транспортер-опосредованного обмена через клеточную мембрану [Borisova, 2017; Borisova, 2016; Chebib, 1999; Shimmura, 2013]. Эти транспортеры завершают синаптическую нейротрансмиссию и запускают обратный захват нейротрансмиттеров. В возбуждающих нейронах глутамат транспортируется в везикулы посредством везикулярных транспортеров глутамата, в то время как в тормозящих нейронах глутамат вначале трансформируется в ГАМК посредством декарбоксилазы глутаминовой кислоты (GAD), а затем транспортируется в везикулы через везикулярные ГАМК транспортеры. После высвобождения оба нейротрансмиттера захватываются высокоаффинными транспортерами и возвращаются к нейронам и близлежащей нейроглии для повторного использования. Следовательно, ГАМК, глутамат и глутамин постоянно сменяют друг друга. Однако при аутизме уровни ферментов, контролирующих цикл глутамина-глутамата-ГАМК, изменяются, и следовательно, в мозге пациента с аутизмом метаболизм глутамина-глутамата-ГАМК с большой вероятностью будет атипичен [Fatemi, 2002; Yip, 2007].
Высокая концентрация глутамата и глутамина (Glx), ГАМК в височной доле, а также высокие уровни глутамата в слуховой коре были непосредственно связаны с тяжестью клинических проявлений РАС [Brown, 2013]. Увеличение уровня глутамата/креатина и Glx в передней поясной коре связано с серьезными нарушениями социального взаимодействия и коммуникации [Egerton, 2012; Tebartz van Elst, 2014]. Согласно гипотезе Fatemi о гипер-глутама- тергическом происхождении аутизма [Fatemi, 2008], низкие уровни фермента GAD и увеличенное количество астроцитов, захватывающих синаптический глутамат и ресинтезирующих глутамин и глутамат, приводит к избытку глутамата в коре головного мозга у пациентов с аутизмом. Чрезвычайно низкие уровни изоформ 65 и 67 kDa GAD у людей с РАС могут объяснять увеличение у пациентов с аутизмом уровня глутамата в тромбоцитах и в крови в целом [El-Ansary, 2017]. Дефицит GAD может быть вызван или связан с нарушениями уровней глутамата/ГАМК или плотности транспортеров/ рецепторов в мозге человека с РАС.
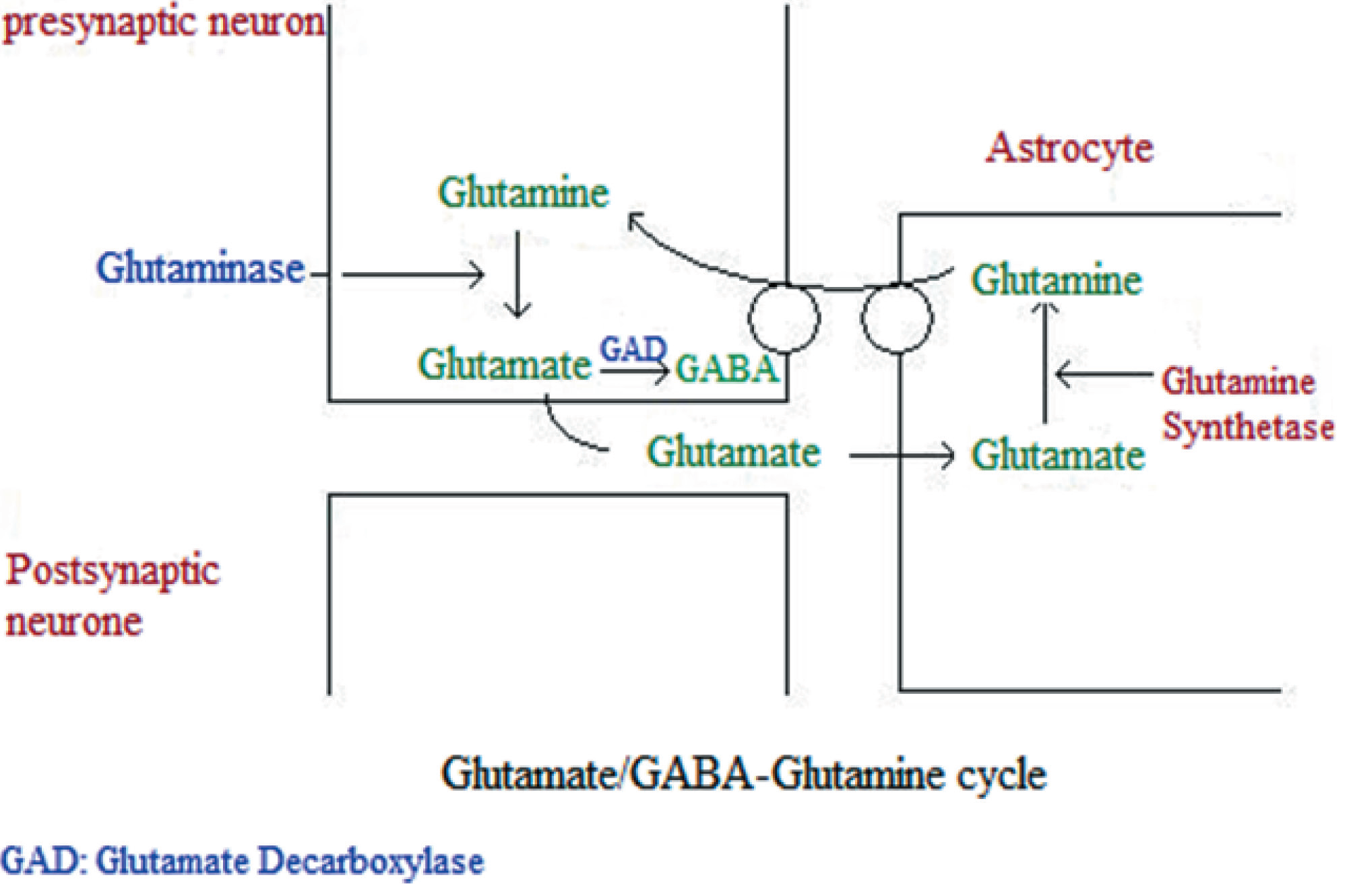
Рис. 1. Цикл глутамат-ГАМК-глутамин
Для конверсии глутамата в глутамин посредством глутаминсинтетазы требуется аммиак, который помогает очистить обе молекулы. Широко известно, что у пациентов с дисфункцией печени и у детей с нарушениями цикла мочевины процесс детоксикации аммиака проходит неэффективно, что приводит к высокой концентрации аммиака и глутамина в мозге [Felipo, 2002]. Согласно Liu et al. [Liu, 2019], у детей с аутизмом наблюдался высокий уровень аммиака в мозге, но низкий уровень содержания глутамина и сывороточного глутамина, а также дисфункция транспортеров глутамата. Эти данные были подтверждены Saleem et al. [Sano, 2009], отметившими повышение уровня аммиака, значительное снижение концентрации мочевины и серьезное увеличение соотношения глутамата/глутамина в плазме пациентов с аутизмом по сравнению с контрольной группой, из чего был сделан вывод о том, что глутамат-глутаминовый цикл у таких пациентов был значительно нарушен. В дополнение, Liu [Liu, 2019] обнаружила группу из 7 индикаторов аминокислот в моче, которые могут отличать образцы мочи детей с РАС и здоровых детей контрольной группы. В совокупности они могут быть связаны с возможным дисбалансом между возбуждающим и тормозящим метаболизмом аминокислот у детей с РАС. Значительно измененные показатели аминокислот в моче могут быть потенциальными диагностическими биомаркерами РАС.
Отметив высокий уровень аммиака в плазме и высокую концентрацию ГАМК в крови и моче мальчика с аутизмом, Cohen [Cohen, 2001] указал на то, что уровни ГАМК в плазме имеют положительную корреляцию с уровнями аммиака в плазме. Dhossche et al. [Dhossche, 2002] обнаружили высокий уровень ГАМК в плазме детей с РАС в возрасте 5—15 лет. Также интересно отметить, что лихорадка приводит к увеличению содержания в спинномозговой жидкости таурина, но к снижению концентрации ГАМК [Liu, 2019].
Было выдвинуто предположение о том, что аммиак, производимый кишечными дрожжевыми грибами Candida albicans, формирует метаболит, функционирующий в мозге человека с РАС подобно ГАМК [Burrus C J, 2012]. Wakefield et al. [Wakefield, 2002] предположили, что кишечные бактерии у детей с аутизмом производят намного больше аммиака, чем может нейтрализовать их ослабленная печень. Например, это может произойти в случае, если произошел чрезмерный рост числа патогенных бактерий в связи с применением антибиотика per orum. Еще одним патогенетическим последствием, связанным с глутаматом, является вакцинация. Hoernlein [Hoernlein, 2019] отметил, что для сохранения вирусного компонента вакцины, в особенности вакцины против кори, свинки, краснухи (MMR), как правило, заключены в оболочку из гидролизованного желатина, который является богатым источником глутамата. В присутствии низкоаффинных транспортеров глутамата, отвечающих за обмен цистина и глутамата между внутренней частью и внешней поверхностью клетки в отношении 1:1, происходит аккумуляция внеклеточного глутамата, что вызывает обструкцию обмена цистина-глутамата, и в результате наблюдается снижение клеточных запасов цистина как главного предшественника серосодержащей аминокислоты и глутатиона. Это может объяснить взаимосвязь между эксайтотоксичностью глутамата и оксидативным стрессом как двумя этиологическими механизмами аутизма [El-Ansary, 2016].
ГАМК-эргическая система и аутизм
Роли ГАМК-эргической системы в аутизме уделяется большое внимание в связи с несколькими факторами. Исследования в области аутизма показали 1) уменьшение количества ГАМК-эргических клеток Пуркинье в мозжечке, особенно в задней доле [Bailey, 1998; Whitney]; 2) снижение уровней активности ключевых синтезирующих ферментов (GAD65 и GAD67) в мозжечке и теменной коре [Fatemi, 2002] и снижение уровней GAD67 в клетках Пуркинье [Yip, 2007]; 3) патологические изменения в глубоких ядрах мозжечка — области мозга, богатой ГАМК-эргическими нейронами, — которые чаще встречаются у мужчин, чем у женщин [Bruchhage, 2018; Kemper, 1998; Mowery, 2015]; 4) снижение плотности рецепторов гамма- аминомасляной кислоты типа А (ГАМК-А) в определенных зонах гиппокампа [Blatt, 2001; Lee, 2014; Varman, 2018]; 5) самая распространенная хромосомная аномалия при аутизме — изменение(я) в хромосоме 15q11-q13 — области, содержащей три гена-кандидата субъединицы рецептора ГАМК-А, связанных с аутизмом [Schroer, 1998; Shao, 2003], ген субъединицы рецептора а5ГАМК-А [Mazzone, 2019]; и 6) повышение содержания ГАМК в плазме детей с аутизмом в возрасте 5—15 лет [El-Ansary, 2016; El-Ansary, 2014].
Рецепторы ГАМК-А — гетеропентамерические ионотропные рецепторы, состоящие из 19 различных субъединиц. Большинство из них имеют две субъединицы а, две субъединицы в и одну субъединицу у или 5 [Olsen, 2009]. Рецепторы ГАМК-А, содержащие субъединицу а5 (а5ГАМК-А), играют важную роль в связи со своей ограниченной дистрибуцией и отличительными физиологическими и фармакологическими характеристиками [Chebib, 1999; Lingford-Hughes, 2002]. Экстрасинаптические рецепторы а5ГАМК-А обладают высоким уровнем экспрессии в гиппокампе и находятся на низком в коре и в гипота - ламусе. Активация данных рецепторов приводит к возникновению стимулирующего ингибирующего тока, который ослабляет возбудимость и синаптическую пластичность [Caraiscos, 2004; Martin]. Рецепторы а5ГАМК-А также играют трофическую роль и регулируют развитие нервных цепей [Lingford-Hughes, 2002].
Интересно отметить, что у мышей с нокаутированным геном Gabra5-/- и с отсутствием гена субъединицы а5 наблюдается множество особенностей, напоминающих симптомы аутизма, например, нарушение социального взаимодействия, аномальные когнитивные функции и функции памяти, а также нарушения сна [Mesbah-Oskui, 2017; Zurek, 2016]. Фактически, самой распространенной вариацией числа копий при аутизме является дупликация участка q11.2-13 хромосомы 15 [Moreno-De-Luca D. et, 2013], кодирующей субъединицы а5, 03 и у3 рецептора ГАМК-А [Mostafa, 2012]. У пациентов с аутизмом были также обнаружены редкие варианты гена, кодирующего субъединицу а5 [Zurek, 2016]. Это позволяет предположить, что дисфункция рецепторов а5 ГАМК-А может приводить к эксайтотоксичности глутамата и к развитию некоторых вариантов РАС. Роль рецепторов ГАМК-А в этиологии аутизма была подтверждена Han et al. [Han, 2014], которые показали, что низкие дозы бензодиазепинов как агонистов рецептора ГАМК-А ведут к увеличению нейротрансмиссии торможения посредством положительной аллостерической модуляции постсинаптических рецепторов ГАМК-А на модели аутизма у грызунов. Это соответствовало улучшению социального взаимодействия и когнитивных способностей, а также сокращению повторяющегося поведения. Напротив, отрицательная аллостерическая модуляция рецепторов ГАМК-А приводила к нарушению социального взаимодействия у мышей дикого типа C57BL/6J и 129SvJ.
Регуляция содержания хлора в нейронах играет важную роль в динамической регуляции ГАМК- эргического ингибирования в процессе пре- и постнатального развития мозга. Данная регуляция зависит в основном от двух котранспортеров кати- он-хлорида (CCC), K+/Cl- котранспортера KCC2 и Na+/K+/Cl- котранспортера NKCC1, активность которого может приводить к снижению или увеличению уровня хлора. Ben-Ari et al. [Ben-Ari, 2014] отметили повышение уровня внутриклеточного хлора (Cl-) и возбуждающей ГАМК на раннем этапе гестации, за которым следовал перинатальный сдвиг от воз - буждения к торможению. Данный механизм можно обнаружить во многих участках мозга различных животных, что говорит о том, что он действует на раннем этапе жизни. В основном, он опосредуется контролируемой во время развития экспрессией генов KCC2 и NKCC1, являющихся экспортером и импортером Cl- соответственно. Несмотря на то, что непосредственная функция рецепторов ГАМК состоит в передаче информации от пресинаптиче- ских нейронов к постсинаптическим, в данном процессе также участвуют некоторые другие факторы. К ним относится разница трансмембранного потенциала постсинаптических дендритов и потенциала реверсии хлорид-ионов. Таким образом, ГАМК может вызывать либо деполяризующие (возбуждающие), либо гиперполяризующие (тормозящие) токи. На это также может повлиять местное распределение больших анионов, таких как глутамат. Существуют три состояния, при которых потенциал реверсии хлорид-ионов ниже, выше или равен мембранному потенциалу покоя. Низкие уровни вну - триклеточного хлора могут вызвать приток более негативно заряженных ионов, что в свою очередь может усилить ингибирующую функцию рецепторов ГАМК-А. Однако при более высоких уровнях хлора потенциал реверсии выше потенциала покоя клетки, что усиливает возбуждающее действие ГАМК. Когда потенциал реверсии хлора равен потенциалу покоя клетки, стимуляция рецепторов ГАМК-А не вызовет результирующий поток хлора и изменение мембранного потенциала. Поскольку возбуждающий потенциал ГАМК и мембранный потенциал покоя находятся в относительной близости, небольшое увеличение Cl- может привести к изменению полярности токов ГАМК-А с тормозящей на возбуждающую, что подчеркивает важность поддержания низкого уровня Cl- [Knoflach , 2016; Lu J.-C, 2014; Raimondo, 2017]. В настоящий момент в исследованиях начинает рассматриваться связь между NKCC1 и KCC2 и этиологией аутизма. Было обнаружено, что мутации в С-терминальном регуляторном домене KCC2 связаны с аутистическими фенотипами [Côme, 2020; Hoernlein, 2019; Merner, 2015], Несколько моделей генетических нарушений, имеющих сильную корреляцию с аутизмом, связаны с изменением соотношения NKCC1/KCC2 [Duarte, 2013]. Воздействие вальпроата (VPA) на крыс в гестационном периоде привело к значительной задержке сдвига ГАМК от возбуждения к торможению [He, 2014]. Оральный прием селективного антагониста NKCC1 буметанида непосредственно перед родами у беременных крыс, подвергавшихся воздействию VPA, приводил к значительному восстановлению эффекта подъема уровня Cl- и возбуждающего сигнального пути ГАМК у новорожденных детенышей, что может препятствовать формированию фенотипов аномального поведения [Rojas, 2014].
Эксайтотоксичность глутамата
как этиологический механизм в аутизме
Эксайтотоксичность глутамата возникает в случае чрезмерной стимуляции рецепторов глутамата (GluRs) избыточным количеством возбуждающего нейротрансмиттера, глутамата, за которым следует повышение межклеточных ионов Ca2+, что в итоге приводит к гибели нейронов [Olloquequi, 2018].
В нескольких исследованиях отмечено, что эксайтотоксичность глутамата, вероятно, является этиологическим механизмом некоторых форм аутизма. GluRs классифицируются как ионотропные рецепторы или метаболические рецепторы (mGluRs) [El-Ansary, 2017; Essa, 2014]. Каждый тип рецепторов состоит из переменной ассоциации субъединиц, определяющей его биофизические и физиологические характеристики. Рецептор N-метил-П-аспартата (NMDAR) — ионотропный GluR, который как правило состоит из тетрадно расположенных субъединиц рецептора подтипов GluN1, GluN2A-D, GluN3A и B. В процессе развития в составе этих субъединиц происходят значительные модификации. В мозге млекопитающих функциональным NMDAR рецептора необходима субъединица GluN1, связанная с одной или большим количеством субъединиц GluN2. Участки связывания магния (Mg2+) внутри NMDAR регулируют его функцию, при этом Mg2+ играет критически важную роль блокатора потенциал-зависимого канала. При деполяризации высвобождается блокада Mg2+, что позволяет продолжение потенциала действия. Чувствительность к блокаде Mg2+ варьирует в зависимости от состава и соотношения субъединиц. Ионы Mg2+ в основном противопоставлены ионам Ca2+; при приеме агонистов NMDA-рецептора можно избежать судорожных припадков, вызванных дефицитом Mg2+ [El-Ansary, 2018а]. Поскольку Mg2+ необходим для многих ферментов мозга, сильное снижение Mg2+ в модели аутизма, вызванного пропионовой кислотой (PPA) у грызунов, недавно было связано с эксайтотоксичностью глутамата как перманентной характеристикой аутизма у ювенильных крыс [El-Ansary, 2018а; Ferguson, 2008; MacFabe, 2007]. В случае дефицита Mg2+ избыточный Ca2+ и глутамат могут вызвать в мозге синаптическую дисфункцию, которая может проявляться как повторяющееся поведение, нарушение социального взаимодействия, судорожные припадки и гиперреактивность, как уже было упомянуто [Al-Suwailem, 2019; Cull-Candy, 2006; Daghestani, 2017].
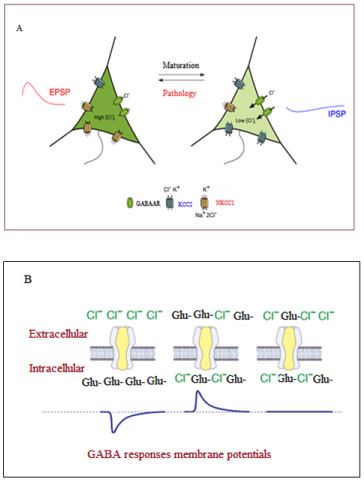
Рис. 2. A: Роль соотношения NKCC1/KCC2 в нейротрансмиссии ГАМК, B: Различные ответные мембранные потенциалы ГАМК
Рецепторы альфа-амино-3-гидрокси-5-метил- 4-изоксазолпропионовой кислоты (AMPAR) — это ионотропные рецепторы, состоящие из субъединиц GluA1-4 (GluR1-4 в старой терминологии). Присутствие субъединицы GluA2 (GluR2) в AMPAR блокирует поступление Ca2+ в нейрон [Biou, 2008]. Чувствительность AMPAR регулируется в зависимости от того, содержат ли они субъединицу GluA2 (GluR2) при транспортировке в нейронную мембрану или нет. В последнем случае AMPAR становятся проницаемыми для Ca2+, и передача AMPAR без GluA2 синаптической единице способствует усилению вызванной глутаматом нейронной активации, которую можно наблюдать в долговременной потенциации (LTP), пластичности, и во время неврологического развития. При патологических состояниях, таких как воспаление, активность AMPAR, не содержащих GluA2, может запустить экс- айтотоксичное повреждение [Ferguson, 2008; Leonoudakis, 2008].
Edfawy et al. [Edfawy, 2019] изучали функциональную роль Gprasp2, гена, связанного с нарушениями психического развития. Этот ген кодирует белок, отвечающий за пост-эндоцитозную организацию рецепторов, сопряженных с G-белком. Они обнаружили, что делеция Gprasp2 приводит к проявлению черт РАС, а также к изменениям синаптической нейротрансмиссии у мышей. Изменение уровней экспрессии Gprasp2 привело к снижению контактной эффективности mGluR5 и изменению дендритной сложности, плотности шиповатых клеток и созревания синапсов. Эти результаты показывают, какую роль играет Gprasp2 в глутаматергических синапсах, и описывают вероятный механизм, посредством которого этот ген может быть связан с аутизмом.
Астроцит-опосредованный клиренс свободного глутамата от синаптической щели в основном происходит посредством двух астроглиальных высокоаффинных транспортеров глутамата — транспортеров возбуждающей аминокислоты 1 и 2 (EAAT1 и 2); в модельных экспериментах на грызунах их классифицируют как транспортер аспартата глутамата (GLAST) и глутамат транспортер-1 (GLT- 1) соответственно. Эти транспортеры используют электрохимические градиенты клеточных мембран в качестве движущих сил для переноса глутамата во внутриклеточное пространство [Anderson, 2000]. Традиционно считалось, что после захвата астроцитами глутамат конвертируется в нетоксичный глутамин посредством глутаминсинтетазы, которая впоследствии высвобождается в межклеточную жидкость и захватывается нейронами для последующего использования с целью рециркуляции глутамата для пополнения пула нейротрансмиттеров [Sibson, 1997]. Однако согласно более современным данным, в некоторых физиологических и патологических условиях астроциты способны высвобождать глутамат с помощью различных механизмов [Canitano, 2017; Vesce, 2007]. Было описано несколько механизмов, связывающих высвобождение глутамата из астроцитов с эксайтотоксичностью, наблюдаемой при аутизме; в основном эти механизмы включают активацию микроглии [Bezzi, 2001; Pascual, 2012; Soni, 2014]. В связи с тем, что EAAT играют ключевую роль в неврологических нарушениях, они являются мишенями для разработки новых стратегий лечения болезней мозга [Grewer, 2005; Soni, 2014]. Согласно одному из последних исследований, бе- та-лактамные антибиотики могут значительно увеличить экспрессию гена EAAT2/GLT-1 на модели аутизма у грызунов [Al-Suwailem, 2019].
Транспортеры высокоаффинного глутамата (L-Glu) способствуют обратному захвату L-Glu в нейроны и клетки нейроглии [Borisova, 2016а]. Эти транспортеры комбинируют захват L-Glu с обменом одного иона H+, одного иона K+ и 3 ионов Na+ [Aoki, 2016]. При аутизме часто говорят о нарушениях транспортеров глутамата и глутамина [Aoki, 2016]. Цистин — это незаменимая аминокислота для биосинтеза восстановленного глутатиона (GSH), для его захвата требуется цистин-глутаматный обменник SLC7A11 [Patel, 2015]. Данный транспортер катализирует захват одной молекулы цистина с высвобождением одной молекулы L-Glu. В связи с его высокой экспрессией в астроцитах, но не в нейронах, нейроны зависят от астроцитов в плане выработки GSH. Когда цистин заходит в астроциты, он формирует трипептид GSH. Синтезированный GSH может высвободиться во внеклеточное пространство, после чего происходит ферментативная реакция, приводящая к формированию цистеина. Затем цистеин захватывается нейронами посредством транспортера SLC1A1 с последующим синтезом GSH. Делеция GSH как патологическая характеристика аутизма может повлиять на способность клеток поглощать свободные радикалы, что делает их подверженными аккумуляции активных форм кислорода (ROS), и может привести к повреждению транспортера L-Glu SLC1A2, особенно в двигательных нейронах. В совокупности с дополнительными изменениями, включая активацию каспаз как про-апоптозных маркеров, это в итоге приводит к гибели.
Ford et al. [Ford, 2019] обнаружили, что сильные аутистические проявления связаны с концентрацией ГАМК+ и с увеличением соотношения глутамат/ ГАМК+ в нижней части правого полушария мозга. Предыдущие исследования также показали, что рост возбуждающей и снижение тормозящей нейротрансмиссии связаны с аутизмом, в особенности с нарушениями в коммуникации [Ford, 2017; Ford, 2017а; Koyama, 2015; Yizhar, 2011]. Эти результаты соответствуют данным, полученным при исследованиях на животных; в этих исследованиях было показано, что нарушения коммуникации, вызванные увеличением соотношения глутамата/ ГАМК+, могут быть уменьшены посредством увеличения тормозящей нейротрансмиссии ГАМК- эргических нейронов [Cellot, 2014; Sgadò, 2013; Yizhar, 2011].
Для понимания роли активации микроглии в дисбалансе возбуждения/торможения при аутизме Koyama и Ikegaya [Lieberman, 2019] выдвинули две различные гипотезы. Широко известно, что в процессе развития в здоровом мозге изначально формируются дополнительные нефизиологические возбуждающие синапсы. Эти ненужные или менее активные синапсы отсекаются микроглией; сохраняются только функционально зрелые синапсы [Bilbo, 2009]. Koyama и Ikegaya выдвинули две гипотезы: 1) при аутизме микроглия может не обнаруживать и не отсекать незрелые синапсы, что приводит к сохранению и избыточному количеству возбуждающих синапсов; и 2) чрезмерно активная микроглия может селективно отсекать ГАМК-эргические синапсы торможения.
Данные гипотезы соответствуют идее о том, что нарушение активации микроглии из-за иммунной стимуляции, например, материнской инфекции в критическом периоде развития, может негативно повлиять на синаптогенез [Bilbo, 2012]. Отсечение микроглией зависит от активности, что говорит о том, что эксайтотоксичная стимуляция в пренатальном или раннем постнатальном периоде может также оказать негативное влияние на архитектуру мозга. Bilbo et al. показали, что активация микроглии мозга на раннем этапе жизни может иметь долго - срочные последствия для функционирования мозга, которые могут проявляться даже во взрослом возрасте [Bilbo, 2012; Biou, 2008].
Дефицит витамина D и дисбаланс
нейротрансмиссии возбуждения/торможения
при аутизме
Известно, что аутизм чаще встречается в регионах с относительно низким воздействием ультрафиолетового излучения, например, в городах и в регионах с большим загрязнением воздуха и высоким количеством осадков [Eyles, 2010]. Исследования на животных подтвердили, что серьезный дефицит витамина D во время беременности оказывает негативное влияние на большое количество белков, отвечающих за развитие мозга, в результате чего у новорожденных животных наблюдаются изменения, аналогичные тем, что мы видим у пациентов с аутизмом [Groves, 2013; Vinkhuyzen, 2018]. Meguid et al [Meguid, 2010] измерили уровень витамина D сыворотки у детей с аутизмом из Египта и сравнили их с результатами контрольной группы. Ученые отметили, что у пациентов с аутизмом уровень витамина D3 был значительно ниже, чем у контрольной группы. Во многих исследованиях было показано, что дефицит витамина D является этиологическим механизмом аутизма [Adams, 2011; El-Ansary, 2018; Fernell, 2015; Gong Z.-L, 2014; Mostafa, 2012; Vinkhuyzen, 2018], и только в двух исследованиях не была обнаружена разница по данному показателю между детьми с аутизмом и контрольной группой [Gürkan, 2014].
Принимая во внимание вышесказанное, рекомендуется назначать добавки с витамином D детям с аутизмом, у которых наблюдается недостаточное содержание данного витамина. Saad et al. [Saleem, 2020] отметили, что добавки с витамином D могут использоваться в качестве стратегии терапии аутизма. В рамках когортного исследования дети с расстройствами аутистического спектра получали добавки витамина D в дозировке 300 МЕ/кг/день; в результате исследования было установлено следующее: 1) дети с конечным уровнем витамина D3 в сыворотке ниже 30 нг/мл не продемонстрировали каких- либо улучшений клинической картины; 2) 31 из 102 детей с конечными уровнями витамина D3 в сыворотке между 30—39 нг/мл показали уменьшение баллов по Оценочной шкале раннего детского аутизма (CARS) на 1,5—4,5 балла; и 3) дети с конечными уровнями витамина D3 в сыворотке выше 40 нг/мл показали снижение по CARS от 3,5 до 6,5 баллов. Предположительно, это говорит о том, что нижняя граница уровня витамина D при терапии аутизма составляет 40 нг/мл, или по крайней мере выше 30 нг/мл. Feng et al. [Ferguson, 2008] показали значительное улучшение состояния людей с РАС и уменьшение симптомов после приема витамина D в течение трех месяцев (при пероральном введении или в форме внутримышечных инъекций), при этом пациенты более младшего возраста лучше отвечали на терапию. Более того, в исследованиях на животных было показано, что витамин D обладает скорее защитным, а не терапевтическим эффектом в случае нейротоксического эффекта, вызванного у крыс пропионовой кислотой, поскольку было обнаружено значительное улучшение измененных уровней интерферона-гамма (IFN-y), серотонина, глутатиона-Б-трансферазы, а также уменьшение повреждения ДНК [Alfawaz, 2014]. Конечно, для изучения взаимосвязи между клиническим ответом на витамин D, долгосрочными последствиями применения добавок, содержащих витамин D и биологическими изменениями у пациентов с аутизмом требуются дополнительные крупномасштабные международные многоцентровые исследования.
Эпидемиологические исследования показали, что дефицит витамина D связан с рядом неврологических и психиатрических нарушений, включая аутизм [Grewer, 2005; Han, 2014; Koyama, 2015; Lee, 2014]. Известно, что дефицит витамина D приводит к значительному снижению уровня глутаматдекарбоксилазы, ключевого фермента ГАМК-эргических интернейронов, а также глутамата и глутамина в мозговой ткани у мышей [Han, 2014]. Таким образом, можно предположить, что эксайтотоксичность глутамата и нарушение цикла глутамат-глутамин-ГАМК при аутизме могут быть скорректированы посредством устранения дефицита витамина D [Wakefield, 2002].
Для проверки данного предположения можно рассмотреть недавнее исследование Krisanova et al. [Lee, 2014], в котором было показано, что дефицит витамина D3 приводит к нарушению синаптической нейротрансмиссии, оказывая воздействие как на Ca2+-зависимые, так и на Ca2+-независимые процессы. Ca2+-зависимое действие дефицита витамина D3 было связано со снижением эксайтотоксичного высвобождения глутамата и ГАМК, предположительно вызванного неправильным функционированием потенциал-зависимых каналов Ca2+. Ca2+- независимое действие дефицита витамина D3 было связано со снижением экспрессии транспортеров глутамата и ГАМК, которое, в свою очередь, при - водит к сокращению обратного захвата глутамата и ГАМК. Это позволило заключить, что дефицит витамина D3 может быть этиологическим механизмом аутизма, приводящим к нарушению экспрессии транспортеров глутамата/ГАМК и дисбалансу возбуждения/торможения.
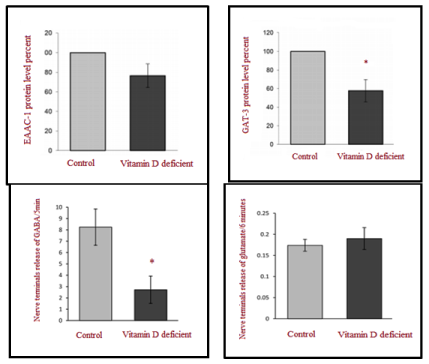
Рис. 3. Экспрессия белка транспортеров EAAC-1 и GAT-3 (повышение) и высвобождение ГАМК и глутамата из синапсов (снижение) у крыс в контрольной группе и группе с дефицитом витамина D3 (на основе работы Krisanova et al. [Lee, 2014])
Роль микробного глутамата и сигнального
пути ГАМК в аутизме
Микробная эндокринология рассматривает роль синтезируемых микроорганизмами веществ, участвующих в нейрохимических процессах, таких как ГАМК, глутамат и серотонин, в качестве общего языка, обеспечивающего взаимодействие микроорганизма и организма хозяина. Понимание взаимоотношений микробиоты кишечника и мозга человека как двунаправленной системы взаимного воздействия, связывающей кишечник и мозг посредством нескольких путей, включая эндокринный, иммунный и нейронный, может сделать возможным использование пробиотиков, синтезирующих вещества, участвующие в нейрохимических процессах, как терапевтической стратегии для лечения аутизма.
Наблюдаемое при аутизме нарушение метилирования может быть связано с дисбалансом соотношения ГАМК/Glu. Нарушение пути метилирования препятствует использованию фолата, распадающегося на глутамат. Цикл Кребса играет ключевую роль в метилировании, и его нарушения могут иметь различный характер, например, дефицит витамина В, присутствие тяжелых металлов и токсинов бактерий или дрожжеподобных грибков рода Candida [Ashwood, 2018]. Известно, что у пациентов с аутизмом наблюдается чрезмерное распространение Candida [Kemper, 1998; Knoflach , 2016]. В случае нарушения метилирования еще больше возрастает важность регуляции уровня глутамата.
Lactobacillus plantarum, L. paracasei, L. lactis, Cory- nebacterium glutamicum, Brevibacterium lactofermentum и B. flavum входят в число бактериальных штаммов, способных синтезировать глутамат [Tebartz van Elst, 2014; Zurek, 2016]. В одном исследовании было обнаружено, что приблизительно 15% штаммов молочнокислых бактерий, изолированных из азиатских ферментированных пищевых продуктов, вырабатывают глутамат [Pokusaeva , 2017]. Грамположительные и грамотрицательные бактерии, такие как E. coli и Pseudomonas, могут вырабатывать ГАМК посредством декарбоксилирования глутамата, катализированного GAD. Было показано, что данный фермент связан с гомеостазом pH и выработкой метаболической энергии [Barrett, 2014].
Исследования показали, что среди микроорганизмов, которые принято считать полезными для здоровья пробиотиками, один штамм Lactobacillus и четыре штамма Bifidobacterium, изолированные из кишечника человека, способны вырабатывать ГАМК [Raimondo, 2017]. Более того, в результате анализа метагеномных данных проекта человеческого микробиома было выдвинуто предположение о том, что гены, кодирующие GAD, могут присутствовать в значительной части микробиоты кишечника человека [Brown, 2013]. ПЦР в реальном времени продемонстрировала снижение количества Lactobacillus и Prevotella в организме самцов и самок мышей с нокаутированным геном Shank3 (генетической модели аутизма) по сравнению с мышами дикого типа. Более того, анализ микробиоты кала и кишечника здоровых самок мышей из контрольной группы показал значительно большее разнообразие родов и видов Lactobacillus по сравнению с микробиотой самцов этой группы. Это позволяет сделать предположение о том, что большее количество и разнообразие Lactobacillus у самок мышей могут служить защитным фактором и объяснением того, почему аутизм чаще встречается у мужчин. В предыдущих исследованиях была показана возможная взаимосвязь между содержанием в организме Lactobacillus, аутичными чертами поведения и ГАМК-эргической функцией. В частности, Bravo et al. [Brown, 2013] определили, что Lactobacillus может регулировать экспрессию рецептора ГАМК в мозге хозяина посредством секреции ГАМК. У мышей с нокаутом гена Shank3 экспрессия рецептора ГАМК особенно сильно нарушена в нескольких зонах мозга, включая гиппокамп, который также является одной из зон, поражаемых, согласно результатам исследования, у пациентов с РАС. Анализ корреляции Пирсона между уровнями L. reuteri, L. brevis, L. ruminis и уровнями рецептора ГАМК в особенности показал, что избыток L. reuteri имеет значительную корреляцию с экспрессией каждой из трех субъединиц рецептора ГАМК (a1GABA-A, a2GABA-A и PIGABA-В).
Терапевтические мишени эксайтотоксичности
глутамата как рациональная стратегия лечения
1. Транспортеры глутамата GLAST/GLT-1
В недавней работе Pajarillo et al. [Pardo, 2005] было отмечено, что регуляция GLAST/GLT-1 может быть нарушена на генетическом, эпигенетическом, транскрипционном или трансляционном уровне, что приводит к высоким уровням внеклеточного глутамата и эксайтотоксич- ности. Соответственно, понимание регуляторных механизмов GLAST/GLT-1 было отмечено как основа разработки терапевтических мишеней для лечения аутизма [Al-Suwailem, 2019; Pardo, 2005]. Лекарственные препараты, такие как бета-лактамные антибиотики, селективные модуляторы рецепторов эстрогенов (SERM), факторы роста, ингибиторы гистондеацетилазы (HDACi) и трансляционные активирующие факторы с заметной эффективностью повышают экспрессию и функционирование GLAST/GLT-1, а также захват глутамата in vitro и in vivo [Pardo, 2005]. Torrez et al. [Tyzio, 2016] указали на нейропротективное действие мемантина (MN), глутаматергическо- го блокатора канала рецептора №метил^-асиартата (NMDAR). Данный препарат мог предотвратить повышение уровней глутамата в спинномозговой жидкости (CSF) и снижение когнитивных способностей у крыс. Его применение способствовало снижению захвата глутамата в гиппокампе и повышению высвобождения белка S100B в CSF в ответ на нейротоксичное воздействие, оказываемое окадаиковой кислотой (OKA).
Этот эффект демонстрирует перспективный защитный механизм взаимодействия нейронов и астроцитов и позволяет отметить, что астроциты могут являться потенциальными мишенями для терапии эксайтотоксичности глутамата.
2. Оксидативный стресс
Оксидативный стресс и сопряженная с ним митохондриальная дисфункция непосредственно связаны с эксайтотоксичностью глутамата при аутизме. Используя множественный регрессивный анализ, El-Ansary [El-Ansary, 2018] показала, что высокие уровни перекисного окисления липидов (ПОЛ) — маркера оксидативного стресса — с одновременным снижением концентрации ферментных и неферментных антиоксидантов (глутатиона, глутатиона/восстановленного глутатиона, тиоредоксина, пероксиредоксинов) были связаны с эксайтотоксичностью глутамата, представленной в форме глутамата, глутамина, соотношения глутамата/глутамина и глутаматдегидрогеназы. На основе этого можно предположить, что оксидативный стресс может быть мишенью для лечения связанных с эксайтотоксичностью глутамата проявлений у пациентов с аутизмом [Angelova, 2018; Smidkova, 2019].
Продукты ПОЛ воздействуют на цепочку транспорта электронов во внутренней митохондриальной мембране, что приводит к потере мембранного потенциала (АТш) и увеличению количества активных форм кислорода (АФК), таких как супероксиданион и пероксид водорода [Merner, 2015]. Несмотря на то, что нейроны обладают сильными механизмами защиты от антиоксидантов, такими как пероксиредоксины, ферменты супероксиддисмутазы (СОД), такие как Cu2+/ 2п2+-СОД (СОД1), Мп2+-СОД (СОД2), глутатионпероксидаза и каталаза (в низкой концентрации), при аутизме их функции серьезно нарушены [Robertson , 2015]. В своей недавней работе Rivero-Segura et al. [Robertson , 2015] оценили возможный антиоксидантный эффект пролактина (PRL), гормона, секретируемого множеством клеток и тканей, включая молочные железы, Т-лимфоциты и гипоталамус. Ученые доказали, что PRL способствует увеличению активности и количества ферментов супероксиддисмутазы (СОД) и снижению концентрации ПОЛ как маркера оксидативного стресса, уровень которого, согласно нескольким исследованиям, у пациентов с аутизмом значительно повышен. Более того, было показано, что PRL предотвращает появление вызванной глутаматом митохондриальной дисфункции и значительно улучшает мембранный потенциал (АТш) дисфункциональных митохондрий, что говорит о его эффективности в качестве метода лечения. В совокупности эти результаты представлены на рис. 4.
3. Инактивация NMDAR и активация рецепторов mGlu
Избыточная активация NMDAR приводит к аккумуляции межклеточного Ca2+ и последующей гибели нейронов [ ]. Многие соединения, включая мемантин и нейростероиды, способны влиять на токсичность глутамата посредством модуляции NMDAR. 20-оксо-5р-прегнан-3а-у1 сульфат (пре- гнанолон сульфат, PAS) — эндогенный нейростероид, ингибирующий токи NMDAR. На глутаматер- гический NMDAR также воздействует множество синтетических нейростероидов [Mead, 2015; Soni, 2014]. Среди проанализированных соединений наилучший NMDAR- опосредованный нейропротективный эффект был выявлен у 3а, 50-прегнанолон глутамат (PAG)—подобного стероидного соединения (рис. 5). Можно предположить, что он может выступать в качестве терапевтического средства для воздействия на связанные с глутаматом проявления аутизма посредством снижения уровня Ca2+, поглощения АФК и предотвращения активации каспазы-3, вызванной глутаматом.
Интересно отметить, что функциональные рецепторы ГАМК-А имеют тенденцию к кластеризации; чрезмерная стимуляция глутаматных NMDA- рецепторов высокими концентрациями глутамата приводит к притоку кальция и потере кластеризации рецепторов ГАМК-А, что негативно влияет на тормозящий эффект ГАМК. Глутамат также может связываться с рецептором mGluR, что приводит к высвобождению хранящегося внутри кальция в цитозоль нейрона. В свою очередь этот кальций может восстановить кластеризацию постсинаптических рецепторов ГАМК-А посредством взаимодействия с протеинкиназой С. Эти результаты показывают, что сигнальный путь глутамата активируется определенными рецепторами и паттернами кальциевой сигнализации, которые препятствуют контролю тормозящих синапсов ГАМК (рис. 6).
4. Активация ГАМК-эргического рецептора
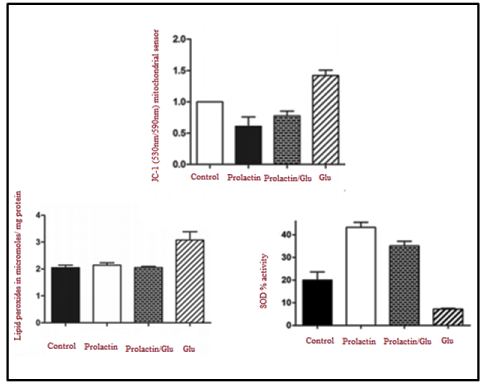
Рис. 4. Терапевтическое воздействие пролактина посредством нейтрализации вызванной глутаматом митохондриальной дисфункции, перекисного окисления липидов и антиоксидантного фермента СОД. JC-1 — датчик потенциала митохондриальной мембраны (ДТт), излучающий флуоресценцию при 590 нм, когда митохондрия поляризована (функциональна), и при 530 нм, когда митохондрия деполяризована (нефункциональна) (на основе работы Rivero-Segura et al. [Robertson , 2015])
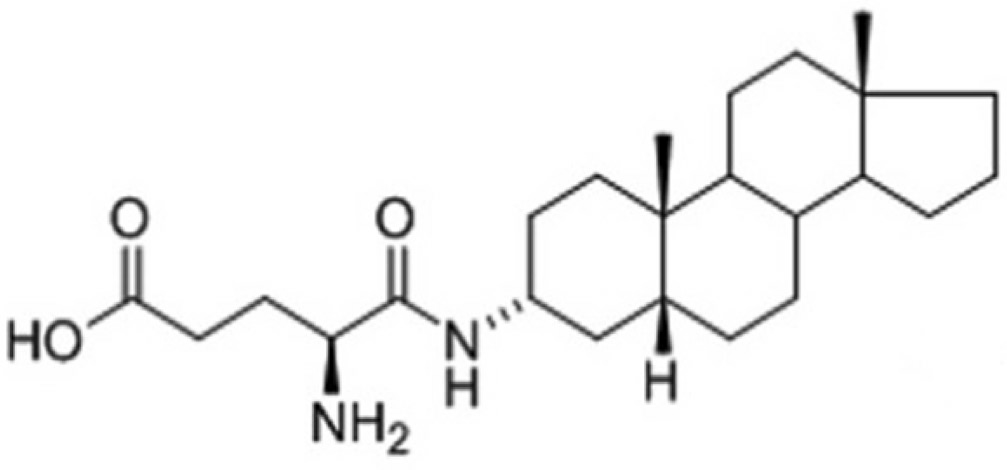
Рис. 5. Химическая структура 3а, 5р-прегнанолон глутамат (PAG)-подобного стероидного соединения, показавшего нейропротективный эффект при вызванной глутаматом эксайтотоксичности
Высвобождение эксайтотоксичных уровней глутамата запускает каскад событий, ведущих к гибели нейронов. Данное явление заключается в дисбалансе между возбуждением и торможением. Mazzone и Nistri [Mead, 2015] выдвинули гипотезу о том, что усиление сети торможения должно предотвращать экс- айтотоксичность и обеспечивать нейропротекцию.
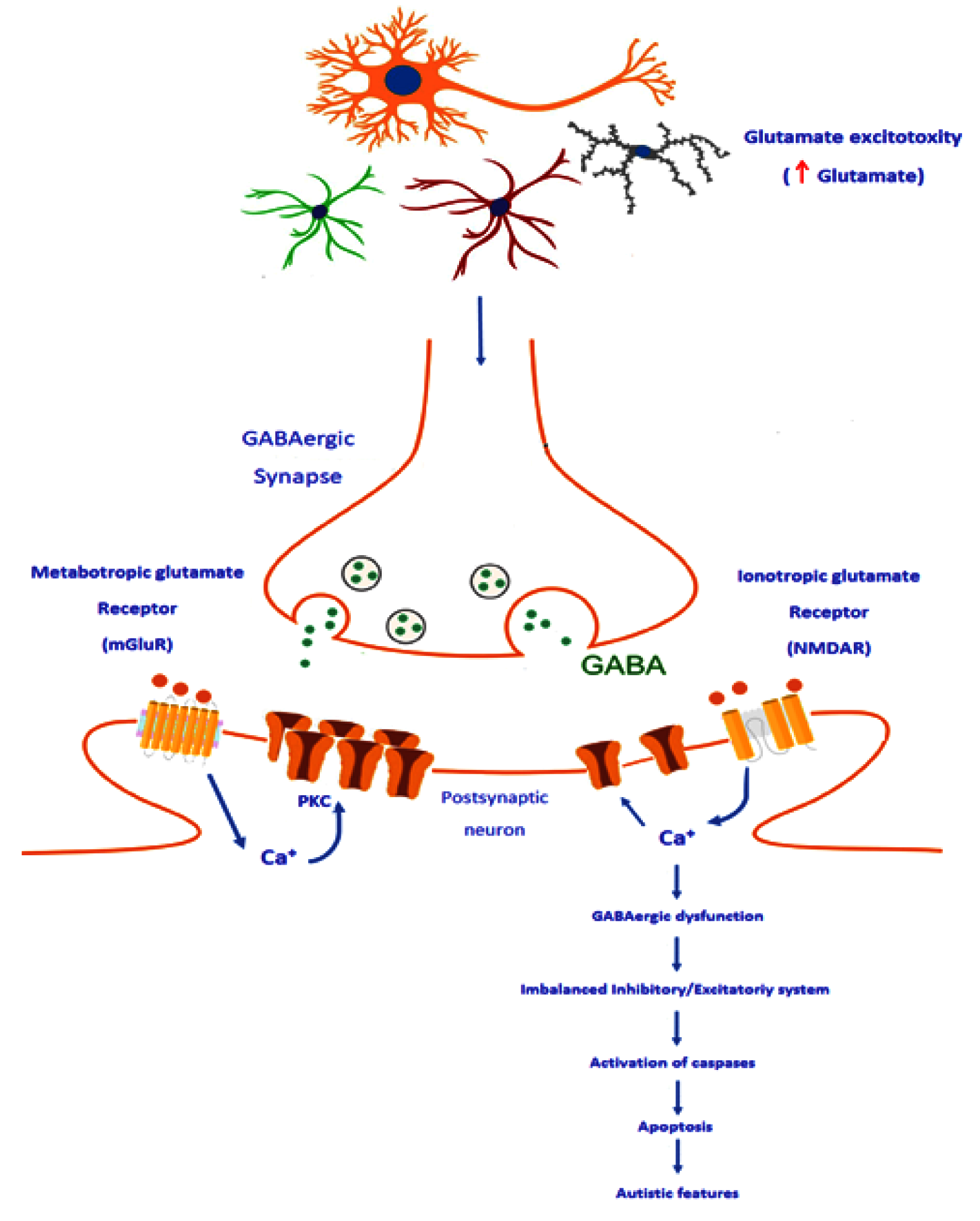
Рис. 6. Предполагаемый механизм дисфункции среди тормозящих связей как этиологический механизм расстройства аутистического спектра посредством потери кластеризации рецепторов ГАМК, вызванной глутаматом, нейротрансмиттером возбуждения. В левой части рисунка показано связывание глутамата с рецептором mGluR, вызывающее высвобождение кальция, хранящегося внутри, в цитозоль нейрона, что приводит к активации протеинкиназы С для поддержания кластеризации и функционирования рецепторов ГАМК-А в постсинаптической мембране. Под воздействием эксайтотоксичности глутамата и понижающей регуляции рецепторов mGluR у пациентов с РАС чрезмерная стимуляция рецепторов NMDA глутаматом приводит к избытку поступления кальция, что вызывает уменьшение кластеризации рецепторов и снижение степени возможности ингибирования нейрона ГАМК. Это может привести к дисбалансу системы возбуждения/торможения, за которым следует активация каспаз (проапоптозные белки), что в итоге приводит к появлению аутистических черт
Они доказали, что высвобождение глутамата, вызванное каинатом, значительно сокращалось посредством аллостерического ГАМК-А-модулятора мидазолама (10 нМ) или агониста ГАМК 4,5,6,7 — тетрагидроазоксазоло[5,4-с]-пиридин-3-олом (THIP; 10 p.M), что приводит к нейропротекции.
Заключение
В данной работе представлены доказательства того, что эксайтотоксичность глутамата является одним из факторов, приводящих к появлению расстройств аутистического спектра; в результате была предложена потенциальная стратегия терапии РАС. Корректировка дисбаланса соотношения глутаматного транспортера/ГАМК у людей с аутизмом может применяться на разных уровнях посредством использования множества мишеней, таких как: 1) активация GLAST/GLT-1 для увеличения обратного захвата глутамата; 2) нейтрализация оксидативного стресса; 3) инактивация NMDAR и активация рецепторов mGlu для надлежащего функционирования рецепторов ГАМК; 4) добавки ГАМК; 5) использование пробиотиков, вырабатывающих ГАМК и GAD, например, Lactobacillus и Bifidobacterial; а также 6) активация ГАМК-эргических рецепторов. В совокупности данные мишени могут помочь понизить уровни глутамата и способствовать надлежащему функционированию рецепторов ГАМК, что делает использование добавок ГАМК успешной и многообещающей стратегией лечения.
Благодаря будущим исследованиям в данном направлении появится возможность создания особых лекарственных препаратов, направленных на эти сигнальные белки, а также модуляции динамики экспрессии NMDAR глутамата, рецепторов и транспортеров mGlu, а также рецепторов ГАМК для терапевтического применения при аутизме.
Рекомендуется проведение дальнейших исследований с использованием оптимальной животной модели и последующее осуществление клинических исследований для доказательства безопасности и эффективности предложенной стратегии с целью повышения успеха трансляционных исследований и их клинического применения в будущем.
Список сокращений:
Рецепторы альфа-амино-3-гидрокси-5-метил- 4-изоксазолпропионовой кислоты (AMPAR); Оценочная шкала раннего детского аутизма (CARS); Котранспортеры катион-хлорида (CCC); Спинномозговая жидкость (CSF); Электроэнцефалограмма (ЭЭГ);Транспортеры возбуждающей аминокислоты 1 (EAAT1); Рецепторы глутамата (GluRs); Транспортер глутамата аспартата (GLAST); Гамма- аминомасляная кислота (ГАМК); Глутамат (Glu); Глутаматдекарбоксилаза (GAD);Глутамат транспортер -1 (GLT-1); Глутамин (Glx); Гамма-аминомасляная кислота типа A (ГАМК-А); Глутатион (GSH); Ингибиторы гистондеацетилазы (HDACi); Интерферон-гамма IFN-y, Котранспортер K+/ Cl- (KCC2); Долговременная потенциация (LTP); Котранспортер Na+/K+/Cl- (NKCC1); Рецептор ^метил-Э-аспартата (NMDAR); Активные формы кислорода (ROS); Ферменты супероксиддисмутазы (СОД);Пролактин (PRL);Пропионовая кислота (PPA); Вальпроат (VPA).
Исследование проведено в соответствии с этическими стандартами Конфликт интересов:
Автор заявляет об отсутствии конфликта интересов.